Services

The mission of the Microbiome Analysis Core is to provide formalized support, at the highest quality standards, for human microbiome-related studies and to foster collaborative initiatives in microbiome research.
The provision of Microbiome Analysis Core services is concentrated around the following aims:
The Microbiome Analysis Core is sustained by a fee-for-service model and grant-incorporated effort levels. The first four hours of consultation are free of charge, and subsequent services are invoiced as detailed below. These rates supports advanced consultation, analysis, administrative tasks, FASRC compute cluster cycles and data storage:
| Category | Hourly rate |
| Internal (33-digit Harvard account code) | $195.00 |
| External academic | $210.00 |
| Industry | $295.00 |
Contact us for consultations, service requests, price quotes, letters of support, or to discuss a collaboration.
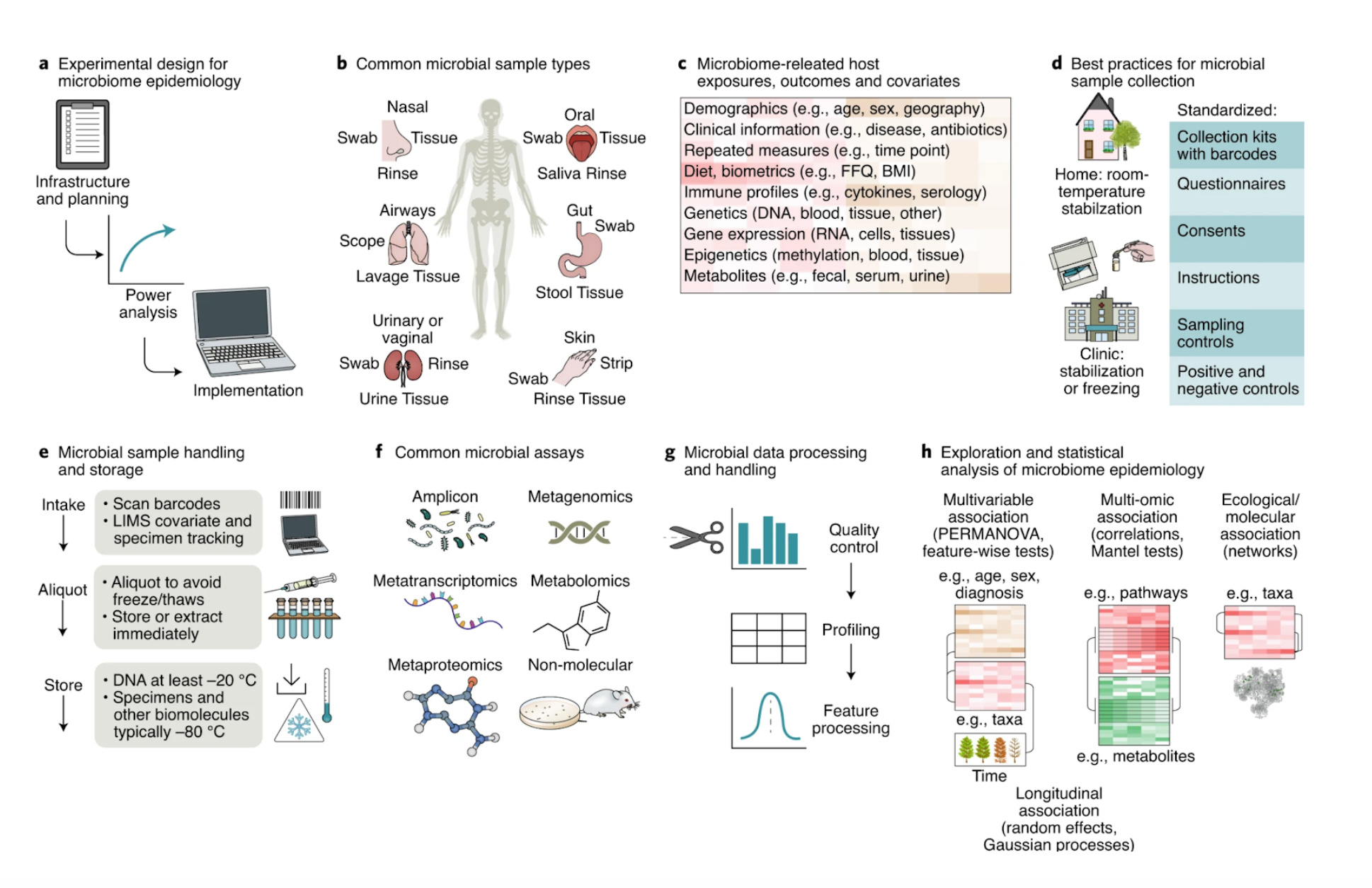
The Microbiome Analysis Core supports microbiome analysis for a variety of molecular data types. A summary of stages to plan for and considerations at each step of microbiome study design and execution. a, Microbiome studies require appropriate physical infrastructure and planning. Power calculations for microbiome studies can be challenging, given the diverse types of measurement possible, their technical variability, and biological factors that can simultaneously influence the microbiome. b, Biospecimens can be collected from diverse sites with varying processing considerations. c, Microbiome analyses are reliant on extensive exposures and phenotypes, as many can affect microbial community state. d, It is critical to have streamlined, hygienic kits, questionnaires, and instructions that are also compatible with downstream sample management and assays. e, Often in combination with controls, specimens can be aliquoted and / or stored long term, or immediately reduced to molecular components. f–h, A wide range of culture-based and culture-independent assays can be applied to population-derived biospecimens (f), after which the resulting molecular data are handled bioinformatically (g) and statistically (h).
Wilkinson, J.E., Franzosa, E.A. et al. A framework for microbiome science in public health. Nature Medicine (2021) PMID: 33820996.
Summary of common workflows of data analysis
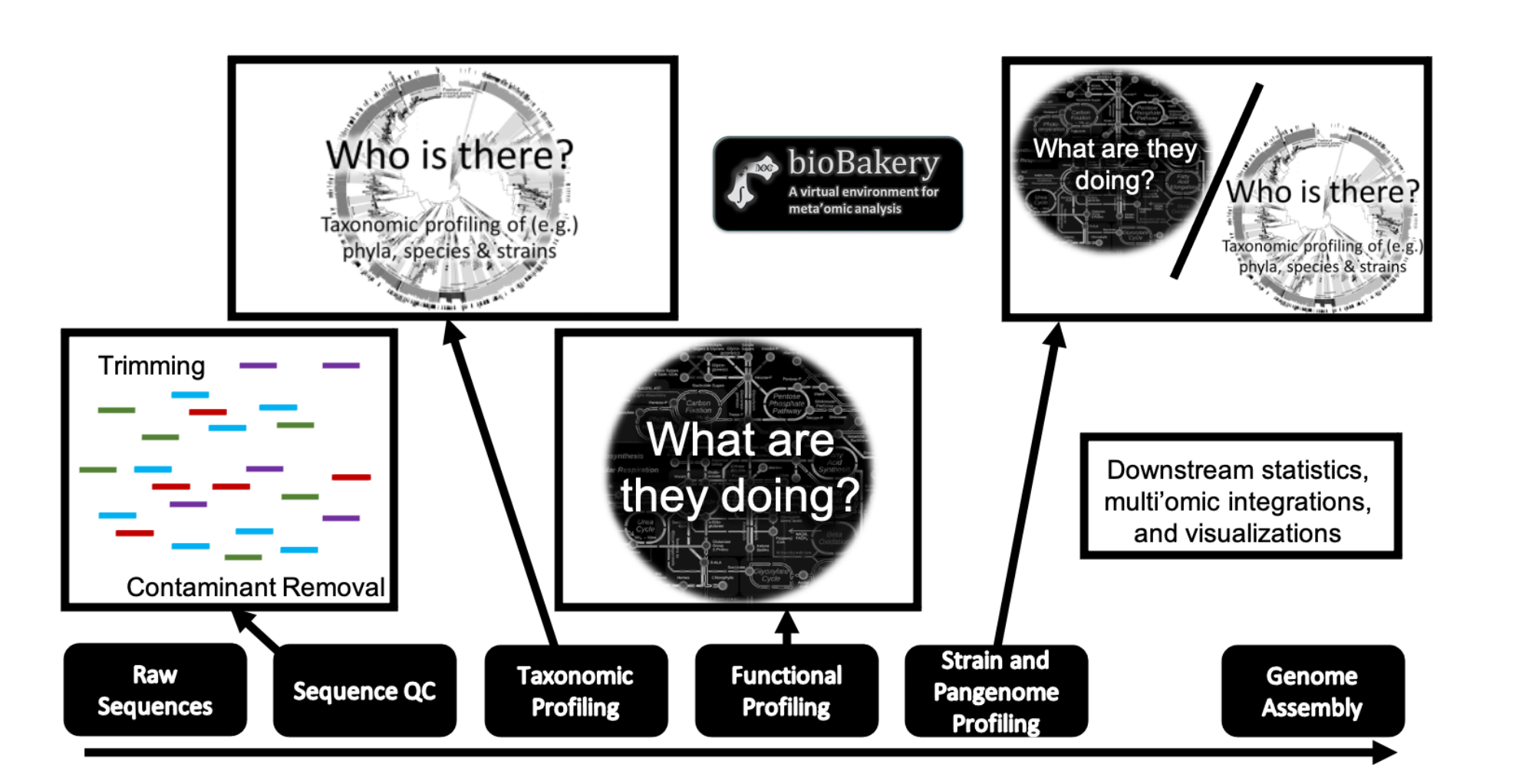
Amplicon (16S rRNA gene / 18S / ITS) or shotgun metagenomic / metatranscriptomic sequencing data is passed through a quality control pipeline using the bioBakery workflows. 16S / 18S / ITS: The amplicon sequence data pipeline provides two approaches, USEARCH / VSEARCH and DADA2 to identify operational taxonomic units (OTUs) and amplicon sequence variants (ASVs), respectively. These taxonomic profiles are then passed to PICRUSt2, which infers gene content and abundance of pathways, to predict the functional profile of the amplicon-resolved community. PICRUSt2 predicted metagenomes are amenable to similar downstream analysis as metagenomes identified from shotgun sequencing data, but with taxonomic resolution limited by 16S / ITS. Microbiome composition (bacteria, archaea, viruses, and eukaryotic microbes) is gleaned from shotgun sequencing data using MetaPhlAn, which resolves taxonomic diversity and abundance at the species and strain level.
Shotgun metagenomes / metatranscriptomes can further be passed through the HUMAnN pipeline. HUMAnN determines conservation and abundance of gene modules (sets of genes related by sequence and function) and biochemical pathways to reveal the metabolic potential of the microbial community.
Data features derived with these algorithms, including gene / pathway presence and abundance, gene expression, microbiome composition, OTUs, ASVs, or peptide identifications from metaproteomics and compound tables from meta-metabolomics, can be integrated with clinical and environmental metadata using LEfSe and MaAsLin 2. LEfSe identifies those data features that are distinct between a pair of metadata values (e.g. differences between two sampling sites, two clinical outcomes, two biochemical markers, two modalities, etc.). MaAsLin 2 extends the functionality of LEfSe to identify associations between data features and multiple metadata variables, which can be discrete and / or continuous and can include time series data.
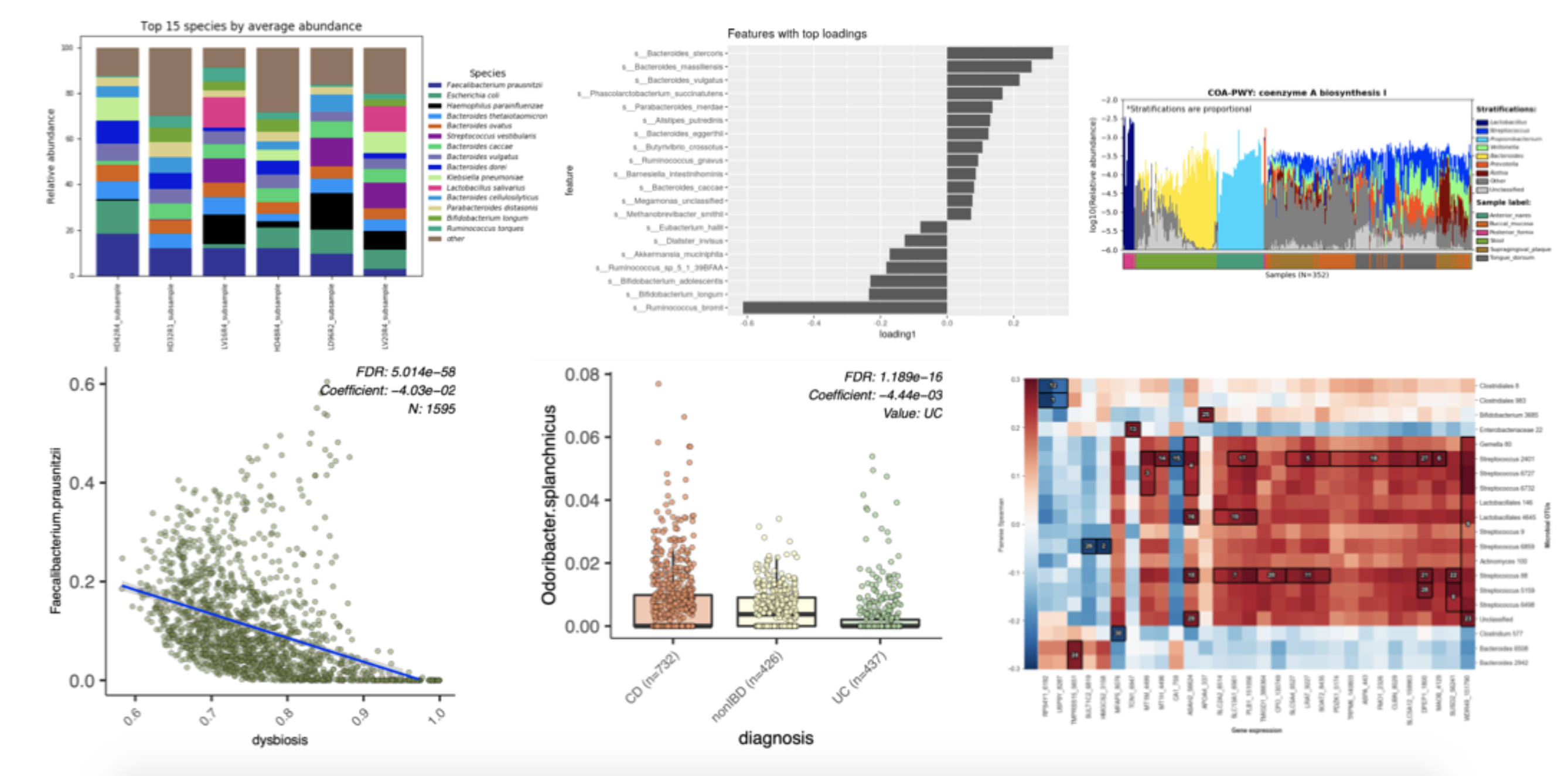
Data Handling
For computing infrastructure, the Microbiome Analysis Core uses the FAS Research Computing cluster. Your meta’omic data is housed on dedicated and regularly backed-up network storage drives. With written consent of the Investigator, data will be removed from our storage after six months of inactivity following completion of collaboration.
Acknowledgement
Co-authorship is not required, but we ask Investigators to adhere to the standard practice of acknowledging Core services in peer-reviewed publications or grant proposals. The Microbiome Analysis Core follows accepted scientific criteria for authorship for statisticians in medical papers (see article), and authorship is discussed with the Investigator at the start of collaboration. Examples of Core contribution that warrant authorship:
-
-
- Major role in study conception and design
- Development of custom analysis methods, tailored specifically for the project
- Biological interpretation of analyzed results
- Contribution of intellectual content to manuscript (not only description of methods used)
-
Intellectual Property
When authorship is not shared, all data prepared and compiled by the Microbiome Analysis Core shall be the property of the Investigator and deemed as works made for hire, upon settlement of invoices for services rendered. Source code for custom built analysis software is not the property of the Investigator, and is reused, distributed, and modified under a Harvard University approved open source license. For provision of services, the Microbiome Analysis Core requires that payment arrangements are made prior to start of collaboration, and that payment is remitted prior to release of results.